Let’s continue where we left off with our earlier exercise by loading in one of our existing hullsets. We’ve already done the hard work of selecting parameters, so we’ll bring in the temporally-dependent hullset using a simple base::file.choose command, which lets you select a file via a browser window. We’ll want to click on the one that has an s of 0.03 in the file name rather than 0. I have the file.choose command commented out for the sake of ‘knitting’ this document (where you cannot interact with the code, so user-based commands will cause it to spit an error. You may see a few more instances of commented out code; just remove the ‘#’ from the front to run them):
#load(file.choose())
load('toni.n5775.s0.03.k15.iso.lhs.01.RData')
Now we have the object we created earlier, called toni.lhs.time.k15. The rest of this exercise will revolve around investigating some of the metrics that we can derive directly from this unique lhs object. Then, we’re going to move on to calculating the isopleth overlap. Finally, we will look into another alternative method, known as Brownian bridges, that also account for some of the unique features of movement data.
We’re going to run this bit of code and then I’ll talk about it while it performs all of the compuations:
library(tlocoh)
## Loading required package: sp
## T-LoCoH for R (version 1.40.04)
## URL: http://tlocoh.r-forge.r-project.org/
## Bug reports: tlocoh@gmail.com
toni.lhs.time.k15 <- lhs.ellipses.add(toni.lhs.time.k15)
## toni.pts5775.k15.s0.03.kmin0
## Calculating enclosing ellipses
## Total time: 1.5 mins
The first metics we are going to examine is called ‘Elongation.’ This is essentially a measure of the eccentircity of the bounding ellipsoid around each hull. For hulls that exhibit high elongation, we can assume the movement summmarized by that hull was directional in nature. On the other hand, hulls that are approximately circular may be more suggestive of foraging or resting behaviors. This is the first example of extracting some behavioral information from our movement trajectories, but it is far from the last example! As you can see from above, we used the tlocoh::lhs.ellipses.add command to do this and on my computer, it took a bit over 3 minutes.
That’s all very nice in theory, but let’s see what it looks like in practice. First, we’ll plot all of the ellipses we just created:
plot(toni.lhs.time.k15, ellipses=TRUE)
That looks pretty neat! We can see ellipses of all different shapes and sizes, but its a bit difficult to make out what is happening with any specfic hull. We can get a bit better resolution on exactly what is happeneing here using a few additional arguments in our plot call:
plot(toni.lhs.time.k15, hulls=T, ellipses=T, allpts=T, nn=T, ptid="auto")
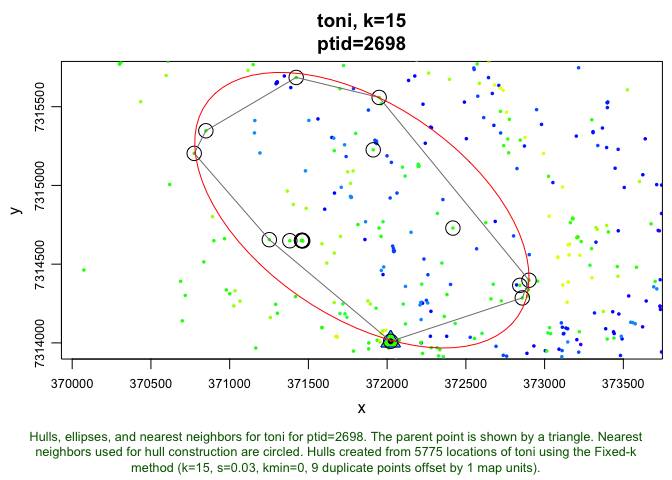
Here we can see the parent point around which the hull is built, the hull itself, and the ellipse that is contructed around it. The circle points that are within the hull are the nearest neighbor points of this particular parent point. One thing you can see is that the hull is clearly not built around only the closest points in terms of spatial proximity (there are a bunch a much closer points). If we call the same line of code again, we will (almost definitely) see a different parent point and its associated neighbors, hull, and ellipse (the ptid="auto" argument leads to the selection of a random point to look at up close).
There are two more important metrics that we will calculate before we look at any in more depth. Both revisitation and visit duration can be derived using the hullset we created. Revisitation represents the number of times that an animal visits the area inside of a given hull whereas duration represents the number of times that an animal was within the hull during a given ‘visit.’ We need to define the inter-visit gap (IVG) period (the unit of time that must pass before another occurrence in the hull is considered a separte visit) in order to calcualte these metrics. Both of these are calculated using the tlocoh::lhs.visit.add. We will use 12 hours as our IVG, which will control for instances where an animal steps out of a hull for a short period before returning to the hull. This must be inputted in terms of seconds, so we will multiply by 3600:
toni.lhs.time.k15 <- lhs.visit.add(toni.lhs.time.k15, ivg=3600*12)
## toni.pts5775.k15.s0.03.kmin0
## 1 of 1. Computing the number of visits in each hull for ivg=43200 (12hs)
If we use the summary command, we can see all of the metrics that we have calculated so far, which should include the ecc (from our ellipses), mnlv.43200 (duration), and nsv.43200 (revistitation):
summary(toni.lhs.time.k15)
## Summary of LoCoH-hullset object: toni.lhs.time.k15
## Created by: T-LoCoH 1.40.4
## [1] toni.pts5775.k15.s0.03.kmin0
## id: toni
## pts: 5775
## dates: 2005-08-23 08:35:00 SAST to 2006-04-23 15:09:00 SAST
## movement: tau=3600 (1hs), vmax=0.9267969, d.bar=173.7452
## hulls: 5775
## dups: 9 (offset by 1 map unit)
## mode: k=15, kmin=0, s=0.03
## metrics: area, ecc, mnlv.43200, nep, nnn, nsv.43200, perim,
## scg.enc.mean, scg.enc.sd, scg.nn.mean, scg.nn.sd, tspan
## hmap: ivg (43200)
## isos: [1] iso.srt-area.iso-q.h5775.i5
## other: ellipses
## created: Sun Dec 3 09:48:16 2017
There they are! Now we’re ready to examine these hull metrics a bit more closely. Earlier, we plotted isopleths based on the density of points (essentially a utilization distribution). What we can do now is create isopleths sorted by something other than density, for example, eccentricity or revisitation. This creates a behavioral map of sorts. Let’s begin by sorting on eccentricity:
toni.lhs.time.k15 <- lhs.iso.add(toni.lhs.time.k15, sort.metric="ecc")
## Merging hulls into isopleths
## toni.pts5775.k15.s0.03.kmin0
## Sorting hulls by ecc descending...Done.
## Unioning hulls
##
|
| | 0%
|
|++++++++++ | 20%
|
|++++++++++++++++++++ | 40%
|
|+++++++++++++++++++++++++++++ | 60%
|
|+++++++++++++++++++++++++++++++++++++++ | 80%
|
|+++++++++++++++++++++++++++++++++++++++++++++++++| 100%
## Total time: 0.8 secs
plot(toni.lhs.time.k15, iso=T, iso.sort.metric="ecc")
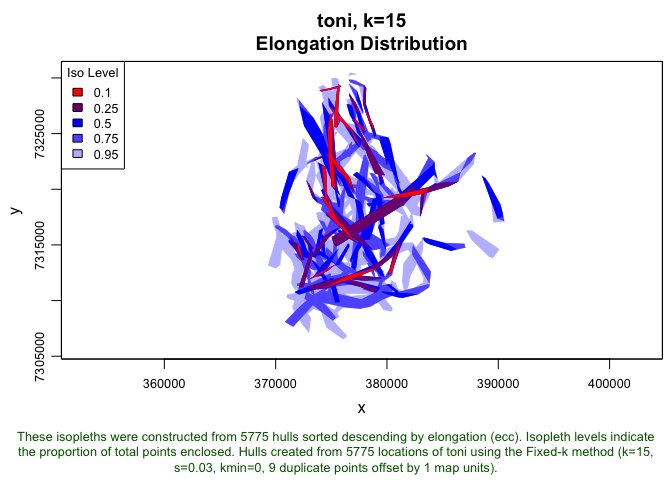
This plot shows some of the areas where Toni moved in the most directed fashion. These could represent efficient routes through the home range (i.e., areas with the least resistance). It should be noted that these home ranges look a bit different from the ones we created earlier because we are sorting by a different metric. Often, this will result in core areas looking a bit more like Swiss cheese than they did when sorting by density. That is to be expected because we are looking, once again, at only the 95-percent isopleth. The 5 percent of points with the lowest eccentricity (which are likely portions of the core area where the animal was not moving in a directional manner) are excluded from this isopleth plot.
Next, lets look at the spatial patterns of revisitation. Let’s look at both the histogram of revisitation, as well as a map of hull parent points colored by revisitation rate (nsv):
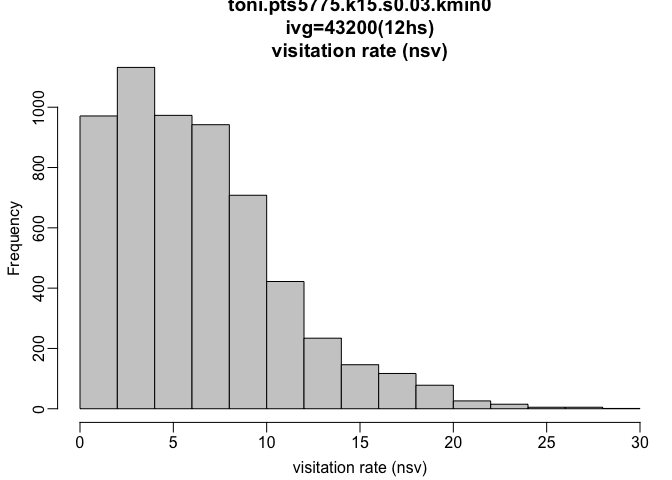
hist(toni.lhs.time.k15, metric="nsv")
plot(toni.lhs.time.k15, hpp=T, hpp.classify="nsv", ivg=3600*12, col.ramp="rainbow")
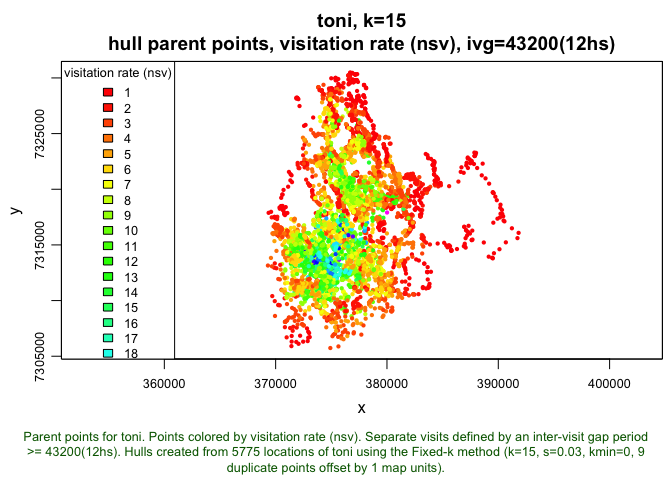
The histogram shows that the majority of hulls were revisted fewer than 10 times, though there are some hulls that are visited almost 30 times! When we look at the plot of the movement track colored by the revistation rate, we can see where these areas of high revisitation are. Unexpectedly, these points are located in the core area, whereas the parent points of hulls with relatively few revisitations are along the periphery. These core areas likely represent regions that contain resources that Toni needed repeatedly. If we wanted to zoom in on a certain portion of the plot, we could use the area of interest command (tlocoh::aoi). Once we type this, we need to click on two points that represent the corners of the box we want to zoom in on (once again, these are commented out because they require active user input, so for the sake of ‘knitting’ they have been removed):
#toni.aoi <- aoi()
#plot(toni.lhs.time.k15, hpp=T, hpp.classify="nsv", col.ramp="rainbow", aoi=toni.aoi)
It could be interesting to lay an NDVI or some other GIS layer under these points to see if these hotspots of revistation correspond to some environmental component.
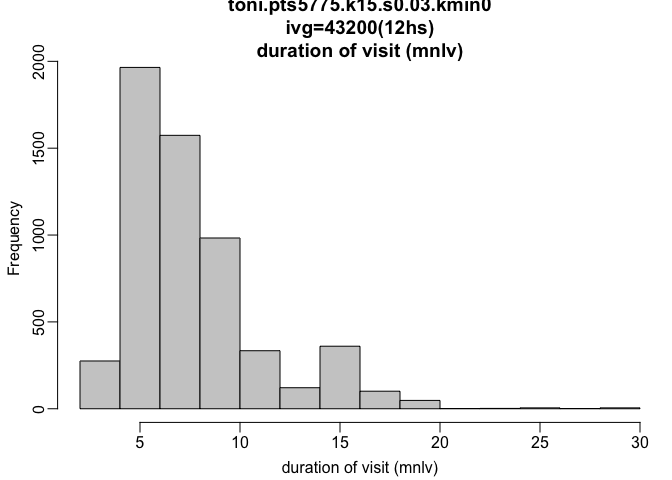
Let’s take a look at duration of each visit now.
hist(toni.lhs.time.k15, metric="mnlv", ivg=3600*12)
plot(toni.lhs.time.k15, hpp=T, hpp.classify="mnlv", col.ramp="rainbow")
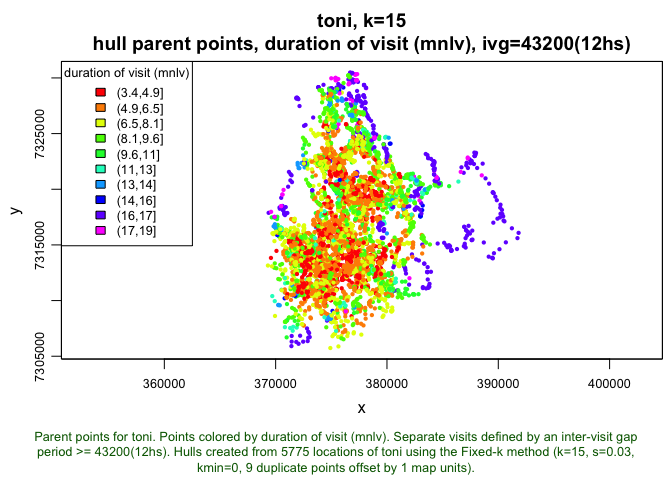
This plot is almost the inverse of the previous. The points that were in the vicinity of the high revisitation areas exhibit lower durations of visit. The latter pattern is often associated with the presence of a watering hole in the home range. The higher values of duration appear to occur along the periphery, but this is a bit misleading; it may actually be an artefact of the fact that we used the k method. Because we used k=15, every hull is constructed from 15 nearest neighors. Furthermore if there are no repeat visits to the area, the hull will consist of 15 temporally contiguous points – a single ‘visit’ with 15 locations. This indicates that the a method may have been better, as it may have accounted for these outlying areas more effectively (i.e., there would be fewer neighbors for those points, and thus, lower durations of visits).
The last thing we’ll look at is a scatterplot of the hull revisitation and duration. We can use this as a map legend. Note how we use a spiral color pattern (instead of rainbow) to help us see where the points fall in the scatterplot, and we’ve also given the scatter plot a black background to make the colors stand out more. When we create the hull metric scatterplot, we save it as an object (hull.scatter) and then feed that object into the plot function for the hull parent points.
hull.scatter <- lhs.plot.scatter(toni.lhs.time.k15, x="nsv", y="mnlv", col="spiral", bg="black")
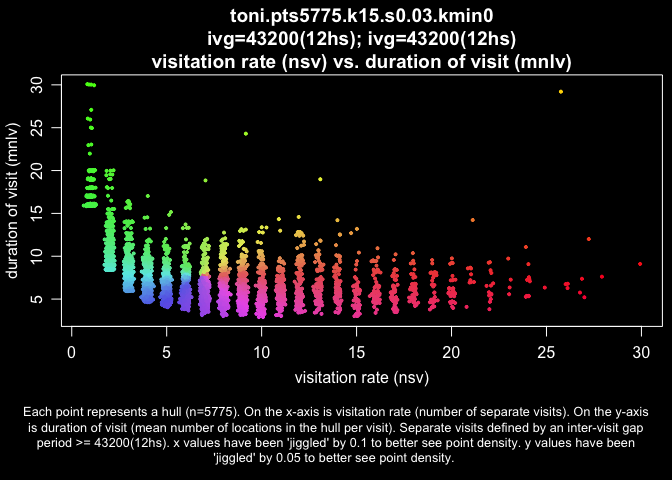
plot(toni.lhs.time.k15, hpp=T, hsp=hull.scatter, hpp.classify="hsp")
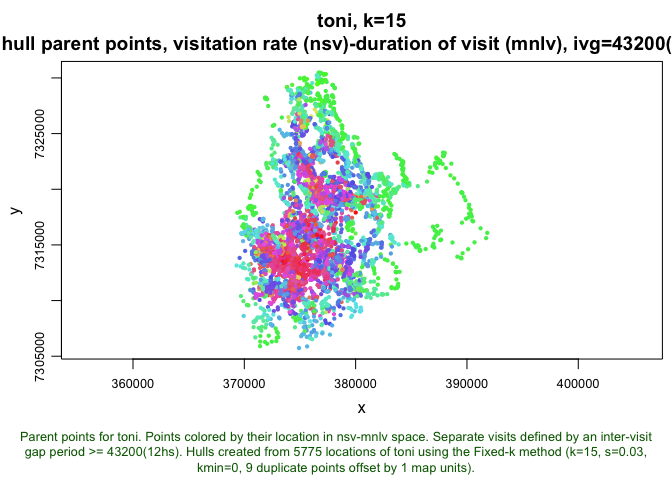
This map illustrates that the measures of time use are not randomly distributed. Rather, these two dimensions of time use do a pretty good job of dividing the landscape into discrete areas.
We can also take a look at a bunch of alternative scatter plots (48 in total) to see if there are any other metrics that might be more helpful for splitting space.
#lhs.plot.scatter.auto(toni.lhs.time.k15)
Now lets use this beautiful hullset that we created for Toni and take a look at its overlap with another one of the buffalo that the team tracked over the same time period, Pepper. Home range overlap is a common analysis for determining the potential for contact between two individuals, which is particularly important when you are interested in disease transmission. To do this, we’ll load in an existing hullset that Andy has created and has placed on the R-Forge server for others to use:
mycon <- url("http://tlocoh.r-forge.r-project.org/pepper.n4571.s0.003.k15.iso.lhs.01.RData")
load(mycon); close(mycon)
This loads in an lhs object called pepper.lhs.k15 which you can now see in your environment. Let’t take a look at this new individual’s movement path:
summary(pepper.lhs.k15)
## Summary of LoCoH-hullset object: pepper.lhs.k15
## Created by: T-LoCoH 1.15
## [1] pepper.pts4571.k15.s0.003.kmin0
## id: pepper
## pts: 4571
## dates: 2005-09-16 17:18:00 SAST to 2006-12-31 16:34:00 SAST
## movement: tau=3600 (1hs), vmax=4.827382, d.bar=158.4384
## hulls: 4571
## dups: 9 (offset by 1 map unit)
## mode: k=15, kmin=0, s=0.003
## metrics: area, nep, nnn, perim, scg.enc.mean, scg.enc.sd,
## scg.nn.mean, scg.nn.sd, tspan
## isos: [1] iso.srt-area.iso-q.h4571.i5
## other: -none-
## created: Tue Mar 11 15:34:50 2014
plot(pepper.lhs.k15, iso=TRUE)
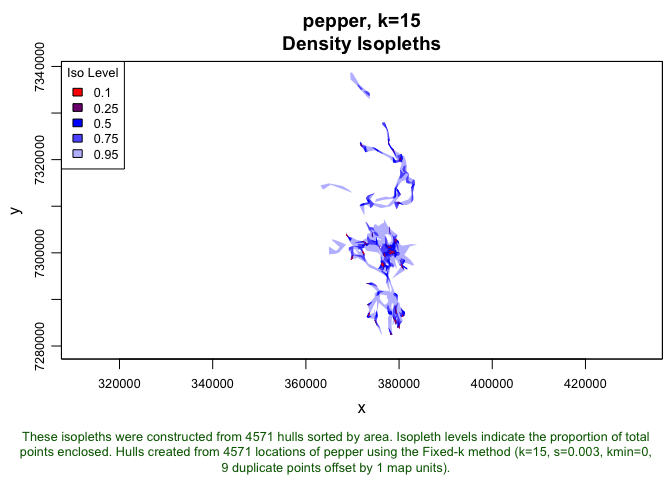
You’ll notice that this object does not have the eccentricity, revisitation, or duration metrics saves like our more complete hullset for Toni, but we could easily add these (though it wont be necessary for this particular analysis):
#pepepr.lhs.k15 <- lhs.ellipses.add(pepper.lhs.k15)
#pepper.lhs.k15 <- lhs.visit.add(pepper.lhs.k15, ivg=3600*12)
The important part for this analysis will be extracting the (density) isopleths from this lhs object. We can isolate this aspect using the tlocoh::isopleths command, which will create a list of SpatialPolygonDataFrame objects:
pepper.isos <- isopleths(pepper.lhs.k15)
We can take a look at what this object looks like by delving into our new pepper.isos object just like any other list:
pepper.isos[[1]]@data
## iso.level area edge.len nep ptp hm.val num.hulls
## 1 0.10 1444531 31071.25 460 0.1006344 85056.01 185
## 2 0.25 6585281 90441.99 1157 0.2531175 171055.91 544
## 3 0.50 24027295 197715.03 2288 0.5005469 350297.19 1179
## 4 0.75 62006071 310103.24 3430 0.7503828 658381.96 2112
## 5 0.95 193837808 367291.09 4344 0.9503391 2342150.10 3512
As we have mentioned previously, the 50% isopleth is often associated with the so-called core area of an animal, whereas the 95% isopleth is frequently used to represent the broader home range. We are going to pull these two polygons from our set (which you can see also includes the 10%, 25%, and 75% isopleths):
pepper.core <- pepper.isos[[1]][ pepper.isos[[1]][["iso.level"]]==0.5, ]
pepper.hr <- pepper.isos[[1]][ pepper.isos[[1]][["iso.level"]]==0.95, ]
plot(pepper.hr, border="blue")
plot(pepper.core, border="red", add=T)

Now we’ll go through the same steps to extract the core area and home range from the Toni lhs that we created:
toni.isos <- isopleths(toni.lhs.time.k15)
toni.core <- toni.isos[[1]][ toni.isos[[1]][["iso.level"]]==0.5, ]
toni.hr <- toni.isos[[1]][ toni.isos[[1]][["iso.level"]]==0.95, ]
plot(pepper.hr, border="blue")
plot(pepper.core, border="red", add=T)
plot(toni.hr, border="green", add=T)
plot(toni.core, border="purple", add=T)

Now we can see there is some potential overlap between these two individuals. We’ll want to be able to quantify that, though, so let’s see if we can extract a value for that region of overlap. We’ll start with the core areas, as these are the areas that both individual use most frequently, which means contact would be most likely in these portions of the ranges. First we’ll plot just these core areas to see if there’s any evidence of overlap at all:
plot(pepper.core, border="red")
plot(toni.core, border="purple", add=T)

Next, we’ll find the area of intersection of the core areas of Toni and Pepper using the gIntersection function from the rgeos package. Then we can visualize these regions on the plot we just made:
library(rgeos)
## rgeos version: 0.3-26, (SVN revision 560)
## GEOS runtime version: 3.6.1-CAPI-1.10.1 r0
## Linking to sp version: 1.2-5
## Polygon checking: TRUE
tp.core.common <- gIntersection(pepper.core, toni.core)
plot(pepper.core, border="red")
plot(toni.core, border="purple", add=T)
plot(tp.core.common, col="black", add=T)

But we wanted an actual metric that we could use to describe this area. Though there is no simple function to determine the polygon area, we can delve into the SpatialPolygon object to get such a value:
tp.core.common@polygons[[1]]@area
## [1] 866424.5
That wasn’t too hard! And now we have an idea of the scale of the overlap between the core areas of these two animals. Though this is the region to the highest probability of the two individuals overlapping, there is still some chance of encounter in the rest of the home range. Let’s use the other polygons that we extracted to define the area of overlap between the 95% isopleths. This will also serve to put this area value that we just extracted into context (i.e., is the core area overlap a large proportion of the home range overlap?)
plot(pepper.hr, border="blue")
plot(toni.hr, border="green", add=T)
tp.hr.common <- gIntersection(pepper.hr, toni.hr)
plot(tp.hr.common, col="black", add=T)
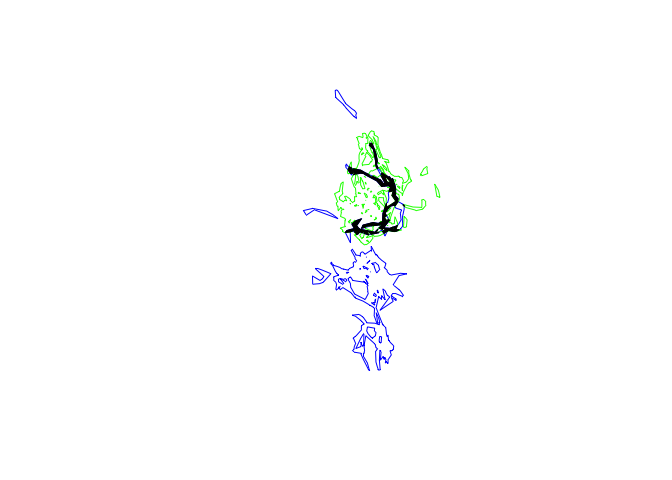
tp.hr.common@polygons[[1]]@area
## [1] 26035939
Clearly this is quite a bit larger than the core overlap area. But just for reference, let’s see what proportion of the overlap is in core areas:
tp.core.common@polygons[[1]]@area / tp.hr.common@polygons[[1]]@area
## [1] 0.03327802
Interesting! Only about 3% of the potential overlap lies in the core areas. That is important to note if we are considering the probability of direct contact between the individuals. From an epidemiological perspective, the home range overlap could be very important for an indirectly transmitted pathogen (such as anthrax) where spatiotemporal overlap is not required, only spatial overlap. In this sense, a much larger area holds potential for transmission.
Next, we’ll take a look at an alternative method for creating a utilization distribution that explicitly incorporates the temporal autocorrelation inherent in movement data. The Brownian Bridge movement model has been a popular means of accounting for some of the uncertainty around each of the sampled position locations (as well as the area in between sampled points).
We’ll start by loading in the fresh toni data and format it appropriately for Brownian Bridge analysis. This will involve loading the data, eliminating duplicate time stamps (something tlocoh did for us automatically), projecting the data into lat-long and then converting it into UTM, and formatting the time as we did earlier. One additional step requires determining the time difference between steps, which we can do quite easily using the base:diff command:
data(toni)
toni <- toni[!duplicated(toni$timestamp.utc),]
toni.sp <- SpatialPoints(toni[ , c("long","lat")], proj4string=CRS("+proj=longlat +ellps=WGS84"))
toni.sp <- spTransform(toni.sp, CRS("+proj=utm +south +zone=36 +ellps=WGS84"))
toni.coords <- data.frame(toni.sp@coords)
colnames(toni.coords) <- c("x", "y")
toni.gmt <- as.POSIXct(toni$timestamp.utc, tz="UTC")
time.diff <- diff(toni.gmt)*60
toni <- toni[-1,]
toni$timelag <- as.numeric(abs(time.diff))
Now on to some more advanced things we’ll need as inputs to create our Brownian bridge layer. We are going to create an empty raster with the resolution that we want our resulting UD to have. To do this, we are going to first extract the extent of Toni’s movements. This can be done easily using the raster::extent command. Then, we’re going to create our own raster that has at least this extent (we will want a buffer just in case the Brownian Bridge highlights areas beyond the bounding box of the movement path). We can do this with the raster::raster command, wherein we will also set the resolution (100 m2 cells) and the projection (to match the rest of our work with Toni). Once we have the raster, we will extract the coordinates of the center points from each cell. The resulting grid will be the basis of the Brownian Bridge.
library(raster)
toni.ext <- extent(toni.sp)
r <- raster(resolution=c(100,100),
xmn = round((toni.ext@xmin - 1000), -2), xmx = round((toni.ext@xmax + 1000), -2),
ymn = round((toni.ext@ymin - 1000), -2), ymx = round((toni.ext@ymax + 1000), -2),
crs = "+proj=utm +south +zone=36 +ellps=WGS84")
r[] <- 0
grid <- coordinates(r)[!is.na(values(r)),]
Now we are ready to implement the Brownian Bridge in the BBMM package using the `BBMM::brownian.bridge command. Unfortunately, this takes quite a while to run, so we are actually going to skip that line and instead, we’ll load in a .tif file of the raster that I created when I initially ran the code on my computer.
library(BBMM)
#toni.BBMM <- brownian.bridge(x=toni.coords$x, y=toni.coords$y, time.lag=toni$timelag, location.error=34, area.grid=grid)
toni.BBMM <- raster('Toni.BBMM.tif')
plot(toni.BBMM)
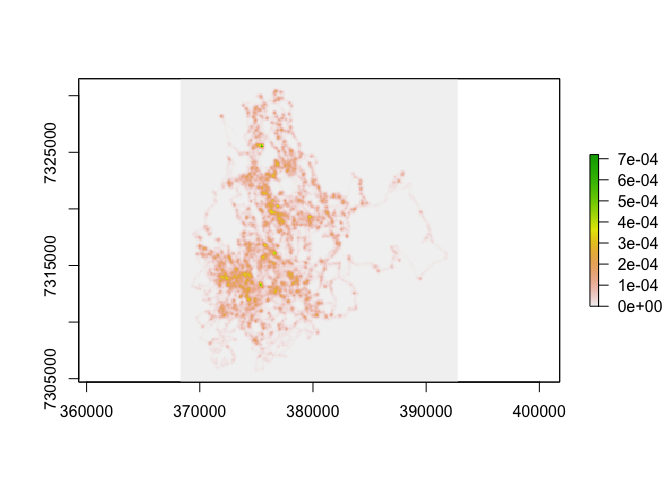
toni.vals <- getValues(toni.BBMM)
Alternatively, we can plot it using the BBMM::bbmm.contour command, but because we did not create a proper bbmm object, we will have to use a long work-around:
#contours <- bbmm.contour(toni.BBMM, levels=c(50, 95), locations=toni, plot=TRUE)
contours <- list(Contour = c("50%", "95%"), Z=c(8.878191e-05, 1.300024e-05))
Next, we can isolate the core area (50%) and home range (95%) for plotting:
library(maptools)
## Checking rgeos availability: TRUE
library(PBSmapping)
##
## -----------------------------------------------------------
## PBS Mapping 2.70.4 -- Copyright (C) 2003-2018 Fisheries and Oceans Canada
##
## PBS Mapping comes with ABSOLUTELY NO WARRANTY;
## for details see the file COPYING.
## This is free software, and you are welcome to redistribute
## it under certain conditions, as outlined in the above file.
##
## A complete user guide 'PBSmapping-UG.pdf' is located at
## /Library/Frameworks/R.framework/Versions/3.4/Resources/library/PBSmapping/doc/PBSmapping-UG.pdf
##
## Packaged on 2017-06-28
## Pacific Biological Station, Nanaimo
##
## All available PBS packages can be found at
## https://github.com/pbs-software
##
## To see demos, type '.PBSfigs()'.
## -----------------------------------------------------------
toni.df <- data.frame(x=data.frame(matrix(grid, ncol=2))[,1], y=data.frame(matrix(grid, ncol=2))[,2], probability=data.frame(matrix(toni.vals, ncol=1))[,1])
bbmm.50 = data.frame(x = toni.df$x, y = toni.df$y, probability = toni.df$probability)
bbmm.50 = bbmm.50[bbmm.50$probability >= contours$Z[1],]
core <- SpatialPixelsDataFrame(points = bbmm.50[c("x", "y")], data=bbmm.50)
## Warning in points2grid(points, tolerance, round): grid has empty column/
## rows in dimension 1
## Warning in points2grid(points, tolerance, round): grid has empty column/
## rows in dimension 2
core <- as(core, "SpatialGridDataFrame")
core <- as(core, "SpatialPolygonsDataFrame")
map.core <- SpatialPolygons2PolySet(core)
map.core <- joinPolys(map.core, operation = 'UNION')
#Set Projection information
map.core <- as.PolySet(map.core, projection = 'UTM', zone = '36')
#Re-assign the PolySet to Spatial Polygons and Polygon ID (PID) to 1
map.core <- PolySet2SpatialPolygons(map.core, close_polys = TRUE)
data.core <- data.frame(PID = 1)
map.core <- SpatialPolygonsDataFrame(map.core, data = data.core)
plot(map.core)
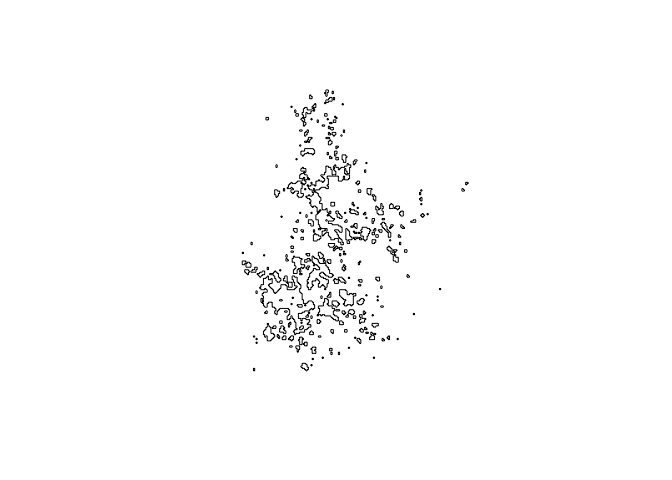
bbmm.95 = data.frame(x = toni.df$x, y = toni.df$y, probability = toni.df$probability)
bbmm.95 = bbmm.95[bbmm.95$probability >= contours$Z[2],]
hr = SpatialPixelsDataFrame(points = bbmm.95[c("x", "y")], data=bbmm.95)
## Warning in points2grid(points, tolerance, round): grid has empty column/
## rows in dimension 1
hr = as(hr, "SpatialGridDataFrame")
hr <- as(hr, "SpatialPolygonsDataFrame")
map.hr <- SpatialPolygons2PolySet(hr)
map.hr <- joinPolys(map.hr, operation = 'UNION')
#Set Projection information
map.hr <- as.PolySet(map.hr, projection = 'UTM', zone = '36')
#Re-assign the PolySet to Spatial Polygons and Polygon ID (PID) to 1
map.hr <- PolySet2SpatialPolygons(map.hr, close_polys = TRUE)
data.hr <- data.frame(PID = 1)
map.hr <- SpatialPolygonsDataFrame(map.hr, data = data.hr)
plot(map.hr)
plot(map.core, col='black', add=TRUE)
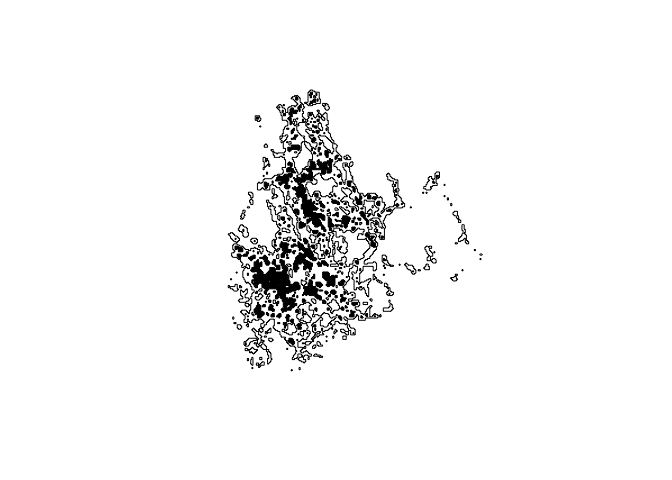
Now we are going to look into one other aspect of the T-LoCoH package that may make various analyses more straightforward or directly comparable. We have seen other forms of utilization distribution (like the BBMM) that take the form of a raster surface, and we can rasterize the isopleths we made earlier in order to compare the T-LoCoH-based UDs to the outputs from alternative methods.
We’ll begin by loading in a few of the packages we will need (raster, tlocoh, and tlocoh.dev which is a version of the package that has some experimental functions). Then we will load in yet another of the buffalo that the Getz Lab tracked in South Africa (named Queen). When we plot this individual, you will see a nice GIS layer plotted beneath it (this is called using the gmap="hybrid" command).
#install.packages('tlocoh.dev', repos="http://R-Forge.R-project.org")
library(tlocoh)
library(tlocoh.dev)
## Loading required package: shiny
## Loading required package: pbapply
## tlocoh.dev contains features under development for T-LoCoH
## Version 1.34.00
## URL: http://tlocoh.r-forge.r-project.org/
## Please send bug reports and feedback to tlocoh@gmail.com
mycon <- url("http://tlocoh.r-forge.r-project.org/queen.n4545.s0.003.k13.lhs.01.RData")
load(mycon); close(mycon)
plot(queen.lhs, allpts=T, gmap="hybrid", cex.allpts=0.2)
## Downloading common background image...Done
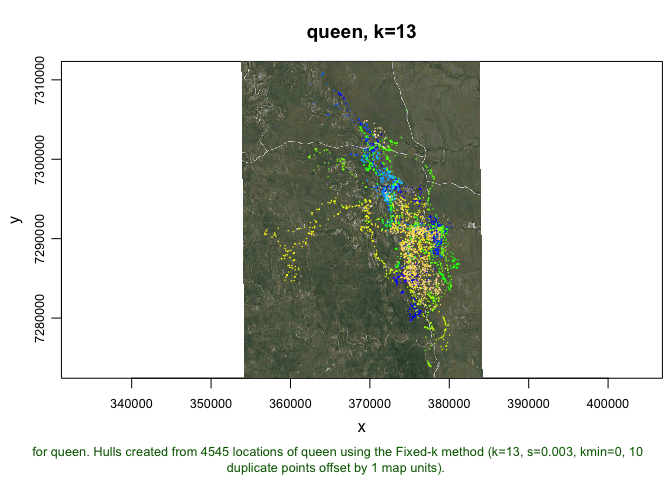
Next, we will build a set of isopleths for Queen, but unlike previous calls, we are going to specify the isopleth levels that we want, and we will select every percentile from 1 to 100 in order to create a near-continuous UD:
queen.lhs <- lhs.iso.add(queen.lhs, iso.levels=1:100/100, status=FALSE)
plot(queen.lhs, iso=TRUE)
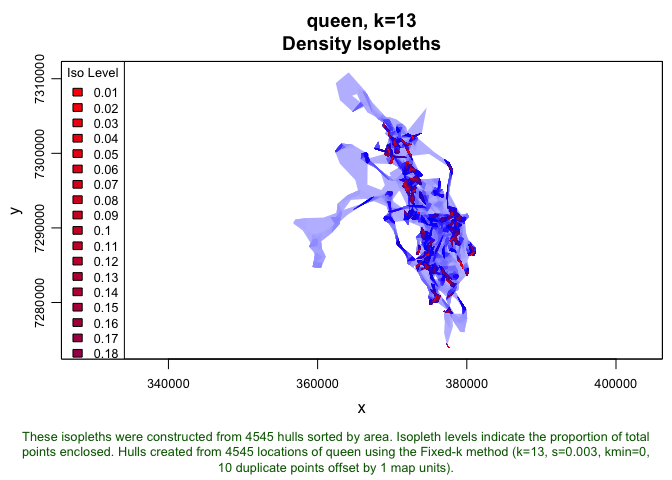
Now, we will rasterize this set of isopleths (this may take a while). We define the resolution of the underlying raster cells using the cell.size argument within the tlocoh::lhs.iso.rast command:
queen.lhs <- lhs.iso.rast(queen.lhs, cell.size=100, status=FALSE)
Then we can verify that the raster that we have created sums to 1 (like all other utilization distributions)
r <- queen.lhs[[1]]$isos[[1]]$rast
sum(getValues(r))
## [1] 1
Once we know that the rasterization was successful, we can plot it:
plot(queen.lhs, rast=T, iso.legend=FALSE, desc=0, title="queen UD")
This layer can now be used in any function that requires a raster input (e.g., adehabitatHR::kerneloverlap which calculates the utilization distribution overlap index (‘UDOI’) or volume of intersection (‘VI’) between home ranges)